
国内有很多的病毒采样管厂家,尤其是疫情以来,病毒采样管的需求持续走高,于国内来说,各大城市都陆续推出了常态化核酸检测,那么需求最大的耗材类产品就是拭子和病毒采样管了,今天在这里单独聊一下病毒采样管的认证应该怎么做。
目前在国内是一类产品,厂家产品生产出之后只需在当地市药监局提出申请拿到产品注册备案和生产备案两个备案即可合规上市销售,相对比较简单,在这里不做赘述,重点聊一下病毒采样管出口欧盟和美国应该怎么做?
病毒采样管出口欧盟需要依据Regulation (EU) 2017/746法规 即体外诊断IVDR法规,属于CLASS A类别产品,厂家只需要完成自我符合性声明+欧代注册即可出口

1、产品测试,IVD产品的测试基本上都是厂家自测,尤其是CLASS A类别产品,主要涉及到的测试市产品本身的性能测试(例如灵敏度、特异性等)
2、依据IVDR法规编写CE技术文件,核心是风险评估和临床评估,需要申请UDI和SRN
3、签署欧代协议,授权欧盟指定代表
4、欧代持完整的技术文件在欧盟当局完成注册
病毒采样管在美国FDA同样属于一类产品,分为灭菌和非灭菌,但无论哪种工艺,最终都是按照一类产品完成FDA注册,只需要识别不同的代码

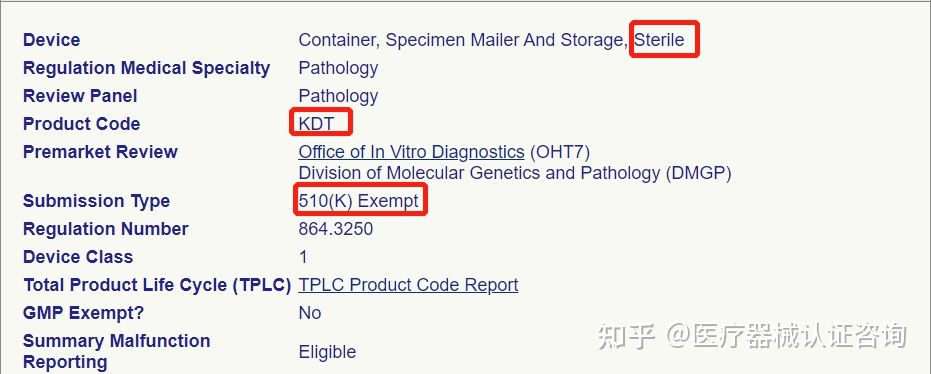
1、企业提供公司基本信息和产品信息
2、申请邓白氏码(目前美国审核没那么严格,没有可以后补)
3、签署美代协议,指定美国代理人
4、完成FDA注册(工厂注册以及企业列名)
需要注意的是豁免510K审核并不意味着豁免测试,只是在注册的过程中无需提交测试报告,后期有可能会被海关或FDA抽查要求提供测试报告,这个企业可视自己情况来决定
其实无论是欧盟CE 还是美国FDA,病毒采样管本质上都不算是一个高风险的产品,但需要注意的是厂家自己对产品的定义,常规的病毒采样管定义就是采集样本的容器,从这个角度来说欧盟和美国都是判定到一类,如果说明书的定义上提到了针对某种病毒采样,比如xinguan,那么就需要重新判断分类和认证途径,视厂家针对的病毒来定义分类,这种做法并不建议,只会增加产品的认证难度和成本,他的本质就只是采集样本的容器而已,无需过多解读!